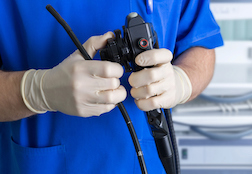
 Endoscopes – and especially duodenoscopes, which are more complex – are reusable devices inserted through the mouth of the patient in order to investigate and diagnose problems with the throat, stomach or intestinal tract. There are specific protocols and processes required to properly clean and sterilize the scopes following use, so that potentially infectious bacteria won’t be transmitted from one patient to another.
Endoscopes – and especially duodenoscopes, which are more complex – are reusable devices inserted through the mouth of the patient in order to investigate and diagnose problems with the throat, stomach or intestinal tract. There are specific protocols and processes required to properly clean and sterilize the scopes following use, so that potentially infectious bacteria won’t be transmitted from one patient to another.It would be cost-prohibitive to dispose of the scopes completely following use, so reusing equipment associated with thorough cleaning is standard procedure.
However – as many an endoscopic infection lawsuit asserts – thorough cleaning and disinfection may not be enough.
Even the FDA admitted, in a warning released two years ago this month, that even the most meticulous cleaning of a duodenoscope prior to disinfection ‘should’ reduce the risk of transmitting and passing along an infection, “but may not entirely eliminate it.”
Olympus Corp. has previously paid a criminal fine and civil settlement for anti-kickback statute (AKS) violations totaling $646 million. The settlement resolved a charge laid by the US Government over alleged conspiracy to violate AKS by the offering of kickbacks to doctors and hospitals in the form of consulting payments, travel, meals, grants and free products such as endoscopes.
However, pundits and commentators opine that government regulators, such as the FDA and those that give the regulator its power, are not blameless. Earlier this month an investigative report released
by the US Government Accountability Office (GAO) suggested that over the past two decades the FDA has approved 25 surgical devices that have the potential to spread undiagnosed cervical cancers in women. The reason for the approvals, according to the GAO report, was that the devices were deemed so similar to older devices, for surgery on the knee and other joints, as an example, that they were approved for use in gynecologic surgeries without rigorous testing. The ramifications of this long-anticipated report are far-reaching.
Furthermore, the report found that holes existed in the agency’s reporting systems meant that reports of deaths or serious injuries related to medical devices made by hospitals and doctors, either didn’t provide alerts to the government, or health regulators didn’t see the incidence reports.
The FDA 510(k) Clearance program is available for any medical device update deemed similar to an existing device – such as an endoscope or duodenoscope – already on the market and performing within specifications. This spares the manufacturer from undertaking the due diligence of an exhaustive clinical trial normally required to approve a product for market, to assure it is safe and effective for the requisite patient cohort.
However – as has been the case with the complexities of duodenoscopes – the design updates approved within the 510(k) framework did not pick up, or so it has been alleged, on unique risks posed by the updated design not necessarily associated with the original design.
The FDA noted in February of 2015 that duodenoscopes have what are known as ‘elevator mechanisms’ that allow the doctor to alter the angle of the device. However, the elevator had the potential to effectively prevent adequate sterilization.
The 510(k) Clearance program was originally envisioned to place promising drugs and devices into the market faster, so patients could benefit more quickly. Critics of the loophole opine that the end result is the movement of potentially unsafe products into the market and into widespread use with the general population without proper vetting, while sponsoring corporations reap the benefit of fast tracked revenue streams.
READ MORE ENDOSCOPE INFECTION LEGAL NEWS
With the FDA already struggling under the weight of job vacancies within the regulator numbering 1,000 or more, the current Administrative hiring freeze coupled with stated objectives to fast track drugs and medical devices could be troubling, say pundits.
The result could provide a continuing pathway for surgical infection lawsuits through the use of devices updated in the absence of detailed testing and trials.